En mi anterior publicación, hable del Albinismo como una de las posibles causas de la heterocromía. Vamos detallar un poco más de que trata esta enfermedad.
¿Qué es el Albinismo por definición?
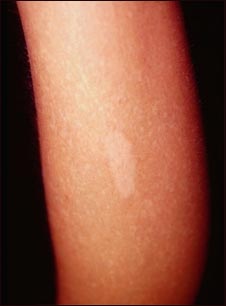
Es un defecto en la producción de melanina (La melanina es una sustancia natural que le da color (pigmento) al cabello, la piel y al iris del ojo, al igual que ayuda a proteger la piel del sol.) que ocasiona una ausencia parcial o total de pigmentación (color) de la piel, el cabello y los ojos.
¿Qué formas tiene de producirse?
Principalmente son dos:
En la primera, o albinismo de tipo 1; los defectos en el metabolismo de la tirosina (La tirosina,abreviada TYR o Y, es uno de los 20 aminoácidos que forman parte de las proteínas), provocan que no se llegue a convertir este aminoácido en melanina. Esto es debido a un defecto genético en la tirosinasa,la enzima encargada de metabolizar la tirosina.
En el segundo caso,o albinismo de tipo 2; se debe a un defecto en el gen "P" y los afectados tienen una pigmentación clara al nacer.
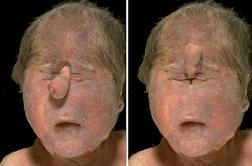
En la forma más grave, llamada albinismo oculocutáneo, las personas afectadas aparecen con cabello, piel y color del iris blanco o rosado, al igual que defectos en la visión. Este tipo de albinismo se hereda a través de un proceso autosómico recesivo (Debe darse, que los dos genes "P" estén afectados por la enfermedad para que se manifieste".
También se da el albinismo en los ojos solamente, llamado albinismo ocular tipo 1 (OA1), y puede ser heredado ya sea en un proceso autosómico recesivo o ligado al cromosoma X. En esta forma, el color de la piel es usualmente normal y el color del ojo puede estar en el rango normal; sin embargo, el examen del ojo muestra que no hay pigmento en la retina (Los pigmentos solo generan un color marrón o negro dependiendo de su concentración, el resto de los colores son efectos ópticos de la luz).
Síndromes y enfermedades ligadas a esta:
-Síndrome de Waardenberg: Es un grupo de afecciones hereditarias caracterizadas por sordera y albinismo parcial (piel, cabello y ojos de color claro). Usualmente un mechón de cabello despigmentado que crece en la frente o ausencia de pigmento en uno o ambos iris (También una de las posibles causas de la heterocromía).
-Síndrome de Chediak-Higashi: Albinismo (falta de pigmento con un diseño repetido en toda la superficie de la piel, pero no es una despigmentación completa.), un brillo plateado en el cabello y ojos de color claro, movimientos espasmódicos del ojo (nistagmo), aumento de las infecciones en los pulmones, la piel y las membranas mucosas.
-Síndrome de Hermansky-Pudlak: Es un trastorno de un gen único heredado de una manera autorecesiva y es una forma de albinismo asociada con un trastorno de sangrado, al igual que con patologías pulmonares e intestinales. Si una persona con albinismo presenta un hematoma o sangrado inusual, se debe pensar en este síndrome.
Curiosidades
Albinismo en Aicuña: En la pequeña y aislada localidad montañosa de Aicuña, en el Oeste de la provincia argentina de La Rioja, se registra un alto porcentaje de albinismo oculocutáneo, un fenómeno producido por la consanguinidad y el régimen de reparto de la tierra.
Elric:Es unos de los personajes importantes en el mundo de la literatura épica-fantástica. Sufre de Albinismo, lo que le produce una gran debilidad física, que contrarresta con medicinas (Drogas).
Elric de Melniboné es un personaje del escritor Michael Moorcock


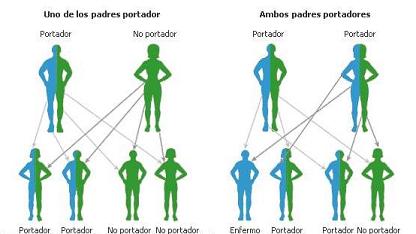

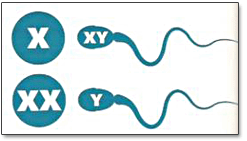
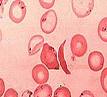 La anemia falciforme es una enfermedad que produce la síntesis de la hemoglobina S, una variante anormal de la hemoglobina. La hemoglobina es una proteína que forma parte de los glóbulos rojos de la sangre y se encarga del transporte de oxígeno a los distintos capilares del cuerpo humano.
La anemia falciforme es una enfermedad que produce la síntesis de la hemoglobina S, una variante anormal de la hemoglobina. La hemoglobina es una proteína que forma parte de los glóbulos rojos de la sangre y se encarga del transporte de oxígeno a los distintos capilares del cuerpo humano.