La evolución del delfín
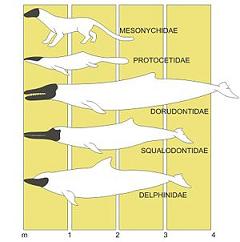
Los delfines, junto con las ballenas y las marsopas, descienden de mamíferos terrestres, más precisamente de los Artiodáctilos. Los esqueletos de los delfines modernos tienen dos pequeños huesos en la pelvis, que indican que alguna vez tuvieron extremidades posteriores.
Un largo proceso evolutivo ha llevado a un pequeño animal terrestre a vivir en el mar hasta transformarse en el actual delfín. Primero tuvo la forma enteramente terrestre hace unos 50 millones de años, por lo tanto, se desarrolla en el transcurso de la Era Terciaria. La forma anfibia se alcanzó después de cinco millones de años, y otros tantos fueron necesarios para que se formara la característica cola con aleta horizontal. Las sucesivas transformaciones se dieron en la dirección de afianzar su especialización, y se refieren al aumento de la masa cerebral y al nacimiento de una forma alargada del morro, con una dentadura diferente.
Estos son los antepasados del delfín actual:
- Mesonychidae:
Antepasado terrestre eocénico (cuarta etapa del periodo Terciario) que vivió hace unos 50 millones de años.
- Protocetidae:
La primera forma acuática conocida, probablemente anfibia, que vivió hace unos 45 millones de años.
- Dorudontidae:
Forma completamente acuática de hace unos 40 millones de años.
- Squalodontidae:
El aspecto se hace similar al del delfín en el Oligoceno (tercera etapa del periodo Terciario), hace unos 25 millones de años.
- Delphinidae:
Los delfines modernos, con el morro en forma de botella, hacen su aparición hace unos 15 millones de años.